Frequently Asked Questions
What is the key idea of Snakemake workflows?
The key idea is very similar to GNU Make. The workflow is determined automatically from top (the files you want) to bottom (the files you have), by applying very general rules with wildcards you give to Snakemake:
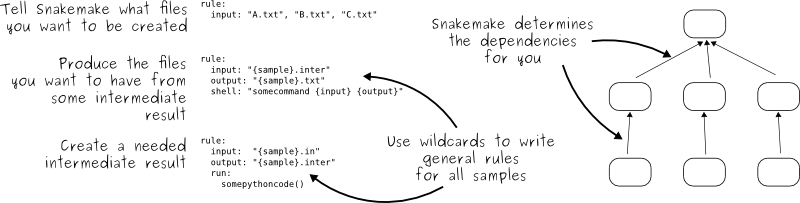
When you start using Snakemake, please make sure to walk through the official tutorial. It is crucial to understand how to properly use the system.
How does Snakemake interpret relative paths?
Relative paths in Snakemake are interpreted depending on their context.
Input, output, log, and benchmark files are considered to be relative to the working directory (either the directory in which you have invoked Snakemake or whatever was specified for
--directoryor theworkdir:directive).Any other directives (e.g.
conda:,include:,script:,notebook:) consider paths to be relative to the Snakefile they are defined in.
If you have to manually specify a file that has to be relative to the currently evaluated Snakefile, you can use workflow.source_path(filepath).
rule read_a_file_relative_to_snakefile:
input:
workflow.source_path("resources/some-file.txt")
output:
"results/some-output.txt"
shell:
"somecommand {input} {output}"
This will in particular also work in combination with modules.
Snakemake does not connect my rules as I have expected, what can I do to debug my dependency structure?
Since dependencies are inferred implicitly, results can sometimes be surprising when little errors are made in filenames or when input functions raise unexpected errors.
For debugging such cases, Snakemake provides the command line flag --debug-dag that leads to printing details each decision that is taken while determining the dependencies.
In addition, it is advisable to check whether certain intermediate files would be created by targeting them individually via the command line.
Finally, it is possible to constrain the rules that are considered for DAG creating via --allowed-rules.
This way, you can easily check rule by rule if it does what you expect.
However, note that --allowed-rules is only meant for debugging.
A workflow should always work fine without it.
My shell command fails with with errors about an “unbound variable”, what’s wrong?
This happens often when calling virtual environments from within Snakemake. Snakemake is using bash strict mode, to ensure e.g. proper error behavior of shell scripts. Unfortunately, virtualenv and some other tools violate bash strict mode. The quick fix for virtualenv is to temporarily deactivate the check for unbound variables
set +u; source /path/to/venv/bin/activate; set -u
For more details on bash strict mode, see the here.
My shell command fails with exit code != 0 from within a pipe, what’s wrong?
Snakemake is using bash strict mode to ensure best practice error reporting in shell commands. This entails the pipefail option, which reports errors from within a pipe to outside. If you don’t want this, e.g., to handle empty output in the pipe, you can disable pipefail via prepending
set +o pipefail;
to your shell command in the problematic rule.
I don’t want Snakemake to detect an error if my shell command exits with an exitcode > 1. What can I do?
Sometimes, tools encode information in exit codes bigger than 1. Snakemake by default treats anything > 0 as an error. Special cases have to be added by yourself. For example, you can write
shell:
"""
set +e
somecommand ...
exitcode=$?
if [ $exitcode -eq 1 ]
then
exit 1
else
exit 0
fi
"""
This way, Snakemake only treats exit code 1 as an error, and thinks that everything else is fine. Note that such tools are an excellent use case for contributing a wrapper.
How do I run my rule on all files of a certain directory?
In Snakemake, similar to GNU Make, the workflow is determined from the top, i.e. from the target files. Imagine you have a directory with files 1.fastq, 2.fastq, 3.fastq, ..., and you want to produce files 1.bam, 2.bam, 3.bam, ... you should specify these as target files, using the ids 1,2,3,.... You could end up with at least two rules like this (or any number of intermediate steps):
IDS = "1 2 3 ...".split() # the list of desired ids
# a pseudo-rule that collects the target files
rule all:
input: expand("otherdir/{id}.bam", id=IDS)
# a general rule using wildcards that does the work
rule:
input: "thedir/{id}.fastq"
output: "otherdir/{id}.bam"
shell: "..."
Snakemake will then go down the line and determine which files it needs from your initial directory.
In order to infer the IDs from present files, Snakemake provides the glob_wildcards function, e.g.
IDS, = glob_wildcards("thedir/{id}.fastq")
The function matches the given pattern against the files present in the filesystem and thereby infers the values for all wildcards in the pattern. A named tuple that contains a list of values for each wildcard is returned. Here, this named tuple has only one item, that is the list of values for the wildcard {id}.
I don’t want expand to use the product of every wildcard, what can I do?
By default the expand function uses itertools.product to create every combination of the supplied wildcards.
Expand takes an optional, second positional argument which can customize how wildcards are combined.
To create the list ["a_1.txt", "b_2.txt", "c_3.txt"], invoke expand as:
expand("{sample}_{id}.txt", zip, sample=["a", "b", "c"], id=["1", "2", "3"])
I don’t want expand to use every wildcard, what can I do?
Sometimes partially expanding wildcards is useful to define inputs which still depend on some wildcards.
Expand takes an optional keyword argument, allow_missing=True, that will format only wildcards which are supplied, leaving others as is.
To create the list ["{sample}_1.txt", "{sample}_2.txt"], invoke expand as:
expand("{sample}_{id}.txt", id=["1", "2"], allow_missing=True)
If the filename contains the wildcard allow_missing, it will be formatted normally:
expand("{allow_missing}.txt", allow_missing=True) returns ["True.txt"].
Snakemake complains about a cyclic dependency or a PeriodicWildcardError. What can I do?
One limitation of Snakemake is that graphs of jobs have to be acyclic (similar to GNU Make). This means, that no path in the graph may be a cycle. Although you might have considered this when designing your workflow, Snakemake sometimes runs into situations where a cyclic dependency cannot be avoided without further information, although the solution seems obvious for the developer. Consider the following example:
rule all:
input:
"a"
rule unzip:
input:
"{sample}.tar.gz"
output:
"{sample}"
shell:
"tar -xf {input}"
If this workflow is executed with
snakemake -n
two things may happen.
If the file
a.tar.gzis present in the filesystem, Snakemake will propose the following (expected and correct) plan:rule a: input: a.tar.gz output: a wildcards: sample=a localrule all: input: a Job counts: count jobs 1 a 1 all 2If the file
a.tar.gzis not present and cannot be created by any other rule than rulea, Snakemake will try to run ruleaagain, with{sample}=a.tar.gz. This would infinitely go on recursively. Snakemake detects this case and produces aPeriodicWildcardError.
In summary, PeriodicWildcardErrors hint to a problem where a rule or a set of rules can be applied to create its own input. If you are lucky, Snakemake can be smart and avoid the error by stopping the recursion if a file exists in the filesystem. Importantly, however, bugs upstream of that rule can manifest as PeriodicWildcardError, although in reality just a file is missing or named differently.
In such cases, it is best to restrict the wildcard of the output file(s), or follow the general rule of putting output files of different rules into unique subfolders of your working directory. This way, you can discover the true source of your error.
Is it possible to pass variable values to the workflow via the command line?
Yes, this is possible. Have a look at Configuration. Previously it was necessary to use environment variables like so: E.g. write
$ SAMPLES="1 2 3 4 5" snakemake
and have in the Snakefile some Python code that reads this environment variable, i.e.
SAMPLES = os.environ.get("SAMPLES", "10 20").split()
I get a NameError with my shell command. Are braces unsupported?
You can use the entire Python format minilanguage in shell commands. Braces in shell commands that are not intended to insert variable values thus have to be escaped by doubling them:
This:
...
shell: "awk '{print $1}' {input}"
becomes:
...
shell: "awk '{{print $1}}' {input}"
Here the double braces are escapes, i.e. there will remain single braces in the final command. In contrast, {input} is replaced with an input filename.
In addition, if your shell command has literal backslashes, \\, you must escape them with a backslash, \\\\. For example:
This:
shell: """printf \">%s\"" {{input}}"""
becomes:
shell: """printf \\">%s\\"" {{input}}"""
How do I incorporate files that do not follow a consistent naming scheme?
The best solution is to have a dictionary that translates a sample id to the inconsistently named files and use a function (see Input functions) to provide an input file like this:
FILENAME = dict(...) # map sample ids to the irregular filenames here
rule:
# use a function as input to delegate to the correct filename
input: lambda wildcards: FILENAME[wildcards.sample]
output: "somefolder/{sample}.csv"
shell: ...
How do I force Snakemake to rerun all jobs from the rule I just edited?
This can be done by invoking Snakemake with the --forcerun or -R flag, followed by the rules that should be re-executed:
$ snakemake -R somerule
This will cause Snakemake to re-run all jobs of that rule and everything downstream (i.e. directly or indirectly depending on the rules output).
How should Snakefiles be formatted?
To ensure readability and consistency, you can format Snakefiles with our tool snakefmt.
Python code gets formatted with black and Snakemake-specific blocks are formatted using similar principles (such as PEP8).
How do I enable syntax highlighting in Vim for Snakefiles?
Instructions for doing this are located here.
Note that you can also format Snakefiles in Vim using snakefmt, with instructions located here!
I want to import some helper functions from another python file. Is that possible?
Yes, from version 2.4.8 on, Snakemake allows to import python modules (and also simple python files) from the same directory where the Snakefile resides.
How can I run Snakemake on a cluster where its main process is not allowed to run on the head node?
This can be achieved by submitting the main Snakemake invocation as a job to the cluster. If it is not allowed to submit a job from a non-head cluster node, you can provide a submit command that goes back to the head node before submitting:
qsub -N PIPE -cwd -j yes python snakemake --cluster "ssh user@headnode_address 'qsub -N pipe_task -j yes -cwd -S /bin/sh ' " -j
This hint was provided by Inti Pedroso.
Can the output of a rule be a symlink?
Yes. As of Snakemake 3.8, output files are removed before running a rule and then touched after the rule completes to ensure they are newer than the input. Symlinks are treated just the same as normal files in this regard, and Snakemake ensures that it only modifies the link and not the target when doing this.
Here is an example where you want to merge N files together, but if N == 1 a symlink will do. This is easier than attempting to implement workflow logic that skips the step entirely. Note the -r flag, supported by modern versions of ln, is useful to achieve correct linking between files in subdirectories.
rule merge_files:
output: "{foo}/all_merged.txt"
input: my_input_func # some function that yields 1 or more files to merge
run:
if len(input) > 1:
shell("cat {input} | sort > {output}")
else:
shell("ln -sr {input} {output}")
Do be careful with symlinks in combination with Step 6: Temporary and protected files. When the original file is deleted, this can cause various errors once the symlink does not point to a valid file any more.
If you get a message like Unable to set utime on symlink .... Your Python build does not support it. this means that Snakemake is unable to properly adjust the modification time of the symlink.
In this case, a workaround is to add the shell command touch -h {output} to the end of the rule.
Can the input of a rule be a symlink?
Yes. In this case, since Snakemake 3.8, one extra consideration is applied. If either the link itself or the target of the link is newer than the output files for the rule then it will trigger the rule to be re-run.
I would like to receive a mail upon snakemake exit. How can this be achieved?
On unix, you can make use of the commonly pre-installed mail command:
snakemake 2> snakemake.log
mail -s "snakemake finished" youremail@provider.com < snakemake.log
In case your administrator does not provide you with a proper configuration of the sendmail framework, you can configure mail to work e.g. via Gmail (see here).
I want to pass variables between rules. Is that possible?
Because of the cluster support and the ability to resume a workflow where you stopped last time, Snakemake in general should be used in a way that information is stored in the output files of your jobs. Sometimes it might though be handy to have a kind of persistent storage for simple values between jobs and rules. Using plain python objects like a global dict for this will not work as each job is run in a separate process by snakemake. What helps here is the PersistentDict from the pytools package. Here is an example of a Snakemake workflow using this facility:
from pytools.persistent_dict import PersistentDict
storage = PersistentDict("mystorage")
rule a:
input: "test.in"
output: "test.out"
run:
myvar = storage.fetch("myvar")
# do stuff
rule b:
output: temp("test.in")
run:
storage.store("myvar", 3.14)
Here, the output rule b has to be temp in order to ensure that myvar is stored in each run of the workflow as rule a relies on it. In other words, the PersistentDict is persistent between the job processes, but not between different runs of this workflow. If you need to conserve information between different runs, use output files for them.
Why do my global variables behave strangely when I run my job on a cluster?
This is closely related to the question above. Any Python code you put outside of a rule definition is normally run once before Snakemake starts to process rules, but on a cluster it is re-run again for each submitted job, because Snakemake implements jobs by re-running itself.
Consider the following…
from mydatabase import get_connection
dbh = get_connection()
latest_parameters = dbh.get_params().latest()
rule a:
input: "{foo}.in"
output: "{foo}.out"
shell: "do_op -params {latest_parameters} {input} {output}"
When run a single machine, you will see a single connection to your database and get a single value for latest_parameters for the duration of the run. On a cluster you will see a connection attempt from the cluster node for each job submitted, regardless of whether it happens to involve rule a or not, and the parameters will be recalculated for each job.
I want to configure the behavior of my shell for all rules. How can that be achieved with Snakemake?
You can set a prefix that will prepended to all shell commands by adding e.g.
shell.prefix("set -o pipefail; ")
to the top of your Snakefile. Make sure that the prefix ends with a semicolon, such that it will not interfere with the subsequent commands. To simulate a bash login shell, you can do the following:
shell.executable("/bin/bash")
shell.prefix("source ~/.bashrc; ")
Some command line arguments like –config cannot be followed by rule or file targets. Is that intended behavior?
This is a limitation of the argparse module, which cannot distinguish between the perhaps next arg of --config and a target.
As a solution, you can put the –config at the end of your invocation, or prepend the target with a single --, i.e.
$ snakemake --config foo=bar -- mytarget
$ snakemake mytarget --config foo=bar
How do I enforce config values given at the command line to be interpreted as strings?
When passing config values like this
$ snakemake --config version=2018_1
Snakemake will first try to interpret the given value as number. Only if that fails, it will interpret the value as string. Here, it does not fail, because the underscore _ is interpreted as thousand separator. In order to ensure that the value is interpreted as string, you have to pass it in quotes. Since bash otherwise automatically removes quotes, you have to also wrap the entire entry into quotes, e.g.:
$ snakemake --config 'version="2018_1"'
How do I make my rule fail if an output file is empty?
Snakemake expects shell commands to behave properly, meaning that failures should cause an exit status other than zero. If a command does not exit with a status other than zero, Snakemake assumes everything worked fine, even if output files are empty. This is because empty output files are also a reasonable tool to indicate progress where no real output was produced. However, sometimes you will have to deal with tools that do not properly report their failure with an exit status. Here, the recommended way is to use bash to check for non-empty output files, e.g.:
rule:
input: ...
output: "my/output/file.txt"
shell: "somecommand {input} {output} && [[ -s {output} ]]"
How does Snakemake lock the working directory?
Per default, Snakemake will lock a working directory by output and input files. Two Snakemake instances that want to create the same output file are not possible. Two instances creating disjoint sets of output files are possible.
With the command line option --nolock, you can disable this mechanism on your own risk. With --unlock, you can be remove a stale lock. Stale locks can appear if your machine is powered off with a running Snakemake instance.
How do I trigger re-runs for rules with updated code or parameters?
Similar to the solution above, you can use
$ snakemake -n -R `snakemake --list-params-changes`
and
$ snakemake -n -R `snakemake --list-code-changes`
Again, the list commands in backticks return the list of output files with changes, which are fed into -R to trigger a re-run.
How do I remove all files created by snakemake, i.e. like make clean
To remove all files created by snakemake as output files to start from scratch, you can use
$ snakemake some_target --delete-all-output
Only files that are output of snakemake rules will be removed, not those that serve as primary inputs to the workflow.
Note that this will only affect the files involved in reaching the specified target(s).
It is strongly advised to first run together with --dry-run to list the files that would be removed without actually deleting anything.
The flag --delete-temp-output can be used in a similar manner to only delete files flagged as temporary.
Why can’t I use the conda directive with a run block?
The run block of a rule (see Snakefiles and Rules) has access to anything defined in the Snakefile, outside of the rule. Hence, it has to share the conda environment with the main Snakemake process. To avoid confusion we therefore disallow the conda directive together with the run block. It is recommended to use the script directive instead (see External scripts).
My workflow is very large, how do I stop Snakemake from printing all this rule/job information in a dry-run?
Indeed, the information for each individual job can slow down a dry-run if there are tens of thousands of jobs.
If you are just interested in the final summary, you can use the --quiet flag to suppress this.
$ snakemake -n --quiet
Git is messing up the modification times of my input files, what can I do?
When you checkout a git repository, the modification times of updated files are set to the time of the checkout.
If you rely on these files as input and output files in your workflow, this can cause trouble.
For example, Snakemake could think that a certain (git-tracked) output has to be re-executed, just because its input has been checked out a bit later.
In such cases, it is advisable to set the file modification dates to the last commit date after an update has been pulled.
One solution is to add the following lines to your .bashrc (or similar):
gitmtim(){
local f
for f; do
touch -d @0`git log --pretty=%at -n1 -- "$f"` "$f"
done
}
gitmodtimes(){
for f in $(git ls-tree -r $(git rev-parse --abbrev-ref HEAD) --name-only); do
gitmtim $f
done
}
(inspired by the answer here).
You can then run gitmodtimes to update the modification times of all tracked files on the current branch to their last commit time in git; BE CAREFUL–this does not account for local changes that have not been committed.
How do I exit a running Snakemake workflow?
There are two ways to exit a currently running workflow.
If you want to kill all running jobs, hit Ctrl+C. Note that when using
--cluster, this will only cancel the main Snakemake process.If you want to stop the scheduling of new jobs and wait for all running jobs to be finished, you can send a TERM signal, e.g., via
killall -TERM snakemake
How can I make use of node-local storage when running cluster jobs?
When running jobs on a cluster you might want to make use of a node-local scratch
directory in order to reduce cluster network traffic and/or get more efficient disk
storage for temporary files. There is currently no way of doing this in Snakemake,
but a possible workaround involves the shadow directive and setting the
--shadow-prefix flag to e.g. /scratch.
rule:
output:
"some_summary_statistics.txt"
shadow: "minimal"
shell:
"""
generate huge_file.csv
summarize huge_file.csv > {output}
"""
The following would then lead to the job being executed in /scratch/shadow/some_unique_hash/, and the
temporary file huge_file.csv could be kept at the compute node.
$ snakemake --shadow-prefix /scratch some_summary_statistics.txt --cluster ...
If you want the input files of your rule to be copied to the node-local scratch directory
instead of just using symbolic links, you can use copy-minimal in the shadow directive.
This is useful for example for benchmarking tools as a black-box.
rule:
input:
"input_file.txt"
output:
file = "output_file.txt",
benchmark = "benchmark_results.txt",
shadow: "copy-minimal"
shell:
"""
/usr/bin/time -v command "{input}" "{output.file}" > "{output.benchmark}"
"""
Executing snakemake as above then leads to the shell script accessing only node-local storage.
How do I access elements of input or output by a variable index?
Assuming you have something like the following rule
rule a: output: expand("test.{i}.out", i=range(20)) run: for i in range(20): shell("echo test > {output[i]}")
Snakemake will fail upon execution with the error 'OutputFiles' object has no attribute 'i'. The reason is that the shell command is using the Python format mini language, which only allows indexing via constants, e.g., output[1], but not via variables. Variables are treated as attribute names instead. The solution is to write
rule a: output: expand("test.{i}.out", i=range(20)) run: for i in range(20): f = output[i] shell("echo test > {f}")
or, more concise in this special case:
rule a: output: expand("test.{i}.out", i=range(20)) run: for f in output: shell("echo test > {f}")
There is a compiler error when installing Snakemake with pip or easy_install, what shall I do?
Snakemake itself is plain Python, hence the compiler error must come from one of the dependencies, like e.g., datrie. You should have a look if maybe you are missing some library or a certain compiler package. If everything seems fine, please report to the upstream developers of the failing dependency.
Note that in general it is recommended to install Snakemake via Conda which gives you precompiled packages and the additional benefit of having automatic software deployment integrated into your workflow execution.
How to enable autocompletion for the zsh shell?
For users of the Z shell (zsh), just run the following (assuming an activated zsh) to activate autocompletion for snakemake:
compdef _gnu_generic snakemake
Example:
Say you have forgotten how to use the various options starting force, just type the partial match i.e. --force which results in a list of all potential hits along with a description:
$snakemake --force**pressing tab**
--force -- Force the execution of the selected target or the
--force-use-threads -- Force threads rather than processes. Helpful if shared
--forceall -- Force the execution of the selected (or the first)
--forcerun -- (TARGET (TARGET ...)), -R (TARGET (TARGET ...))
To activate this autocompletion permanently, put this line in ~/.zshrc.
Here is some further reading.
How can I avoid system /tmp to be used when combining apptainer and conda?
When using both apptainer and conda the idea is that inside the apptainer container the conda environment is being installed.
Some apptainer instances are set to share the system /tmp with the containers.
This can lead to unexpected behaviour where the system /tmp gets full.
To stop this behaviour you’d have to run apptainer with the --contain option.