Basics: An example workflow¶
Please make sure that you have activated the environment we created before, and that you have an open terminal in the working directory you have created.
A Snakemake workflow is defined by specifying rules in a Snakefile. Rules decompose the workflow into small steps (e.g., the application of a single tool) by specifying how to create sets of output files from sets of input files. Snakemake automatically determines the dependencies between the rules by matching file names.
The Snakemake language extends the Python language, adding syntactic structures for rule definition and additional controls. All added syntactic structures begin with a keyword followed by a code block that is either in the same line or indented and consisting of multiple lines. The resulting syntax resembles that of original Python constructs.
In the following, we will introduce the Snakemake syntax by creating an example workflow. The workflow will map sequencing reads to a reference genome and call variants on the mapped reads.
Step 1: Mapping reads¶
Our first Snakemake rule maps reads of a given sample to a given reference genome.
In the working directory, create a new file called Snakefile with an editor of your choice.
We propose to use the Atom editor, since it provides out-of-the-box syntax highlighting for Snakemake.
In the Snakefile, define the following rule:
rule bwa_map:
input:
"data/genome.fa",
"data/samples/A.fastq"
output:
"mapped_reads/A.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
A Snakemake rule has a name (here bwa_map) and a number of directives, here input, output and shell.
The input and output directives are followed by lists of files that are expected to be used or created by the rule.
In the simplest case, these are just explicit Python strings.
The shell directive is followed by a Python string containing the shell command to execute.
In the shell command string, we can refer to elements of the rule via braces notation (similar to the Python format function).
Here, we refer to the output file by specifying {output} and to the input files by specifying {input}.
Since the rule has multiple input files, Snakemake will concatenate them separated by a whitespace.
In other words, Snakemake will replace {input} with data/genome.fa data/samples/A.fastq before executing the command.
The shell command invokes bwa mem with reference genome and reads, and pipes the output into samtools which creates a compressed BAM file containing the alignments.
The output of samtools is piped into the output file defined by the rule.
When a workflow is executed, Snakemake tries to generate given target files. Target files can be specified via the command line. By executing
$ snakemake -np mapped_reads/A.bam
in the working directory containing the Snakefile, we tell Snakemake to generate the target file mapped_reads/A.bam.
Since we used the -n (or --dryrun) flag, Snakemake will only show the execution plan instead of actually perform the steps.
The -p flag instructs Snakemake to also print the resulting shell command for illustration.
To generate the target files, Snakemake applies the rules given in the Snakefile in a top-down way.
The application of a rule to generate a set of output files is called job.
For each input file of a job, Snakemake again (i.e. recursively) determines rules that can be applied to generate it.
This yields a directed acyclic graph (DAG) of jobs where the edges represent dependencies.
So far, we only have a single rule, and the DAG of jobs consists of a single node.
Nevertheless, we can execute our workflow with
$ snakemake mapped_reads/A.bam
Note that, after completion of above command, Snakemake will not try to create mapped_reads/A.bam again, because it is already present in the file system.
Snakemake only re-runs jobs if one of the input files is newer than one of the output files or one of the input files will be updated by another job.
Step 2: Generalizing the read mapping rule¶
Obviously, the rule will only work for a single sample with reads in the file data/samples/A.fastq.
However, Snakemake allows to generalize rules by using named wildcards.
Simply replace the A in the second input file and in the output file with the wildcard {sample}, leading to
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
When Snakemake determines that this rule can be applied to generate a target file by replacing the wildcard {sample} in the output file with an appropriate value, it will propagate that value to all occurrences of {sample} in the input files and thereby determine the necessary input for the resulting job.
Note that you can have multiple wildcards in your file paths, however, to avoid conflicts with other jobs of the same rule, all output files of a rule have to contain exactly the same wildcards.
When executing
$ snakemake -np mapped_reads/B.bam
Snakemake will determine that the rule bwa_map can be applied to generate the target file by replacing the wildcard {sample} with the value B.
In the output of the dry-run, you will see how the wildcard value is propagated to the input files and all filenames in the shell command.
You can also specify multiple targets, e.g.:
$ snakemake -np mapped_reads/A.bam mapped_reads/B.bam
Some Bash magic can make this particularly handy. For example, you can alternatively compose our multiple targets in a single pass via
$ snakemake -np mapped_reads/{A,B}.bam
Note that this is not a special Snakemake syntax. Bash is just expanding the given path into two, one for each element of the set {A,B}.
In both cases, you will see that Snakemake only proposes to create the output file mapped_reads/B.bam.
This is because you already executed the workflow before (see the previous step) and no input file is newer than the output file mapped_reads/A.bam.
You can update the file modification date of the input file
data/samples/A.fastq via
$ touch data/samples/A.fastq
and see how Snakemake wants to re-run the job to create the file mapped_reads/A.bam by executing
$ snakemake -np mapped_reads/A.bam mapped_reads/B.bam
Step 3: Sorting read alignments¶
For later steps, we need the read alignments in the BAM files to be sorted.
This can be achieved with the samtools command.
We add the following rule beneath the bwa_map rule:
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
This rule will take the input file from the mapped_reads directory and store a sorted version in the sorted_reads directory.
Note that Snakemake automatically creates missing directories before jobs are executed.
For sorting, samtools requires a prefix specified with the flag -T.
Here, we need the value of the wildcard sample.
Snakemake allows to access wildcards in the shell command via the wildcards object that has an attribute with the value for each wildcard.
When issuing
$ snakemake -np sorted_reads/B.bam
you will see how Snakemake wants to run first the rule bwa_map and then the rule samtools_sort to create the desired target file:
as mentioned before, the dependencies are resolved automatically by matching file names.
Step 4: Indexing read alignments and visualizing the DAG of jobs¶
Next, we need to index the sorted read alignments for random access. This can be done with the following rule:
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
Having three steps already, it is a good time to take a closer look at the resulting DAG of jobs. By executing
$ snakemake --dag sorted_reads/{A,B}.bam.bai | dot -Tsvg > dag.svg
we create a visualization of the DAG using the dot command provided by Graphviz.
For the given target files, Snakemake specifies the DAG in the dot language and pipes it into the dot command, which renders the definition into SVG format.
The rendered DAG is piped into the file dag.svg and will look similar to this:
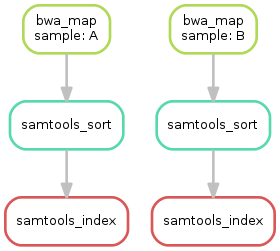
The DAG contains a node for each job and edges representing the dependencies. Jobs that don’t need to be run because their output is up-to-date are dashed. For rules with wildcards, the value of the wildcard for the particular job is displayed in the job node.
Exercise¶
- Run parts of the workflow using different targets. Recreate the DAG and see how different rules become dashed because their output is present and up-to-date.
Step 5: Calling genomic variants¶
The next step in our workflow will aggregate the aligned reads from all samples and jointly call genomic variants on them. Snakemake provides a helper function for collecting input files. With
expand("sorted_reads/{sample}.bam", sample=SAMPLES)
we obtain a list of files where the given pattern "sorted_reads/{sample}.bam" was formatted with the values in the given list of samples SAMPLES, i.e.
["sorted_reads/A.bam", "sorted_reads/B.bam"]
The function is particularly useful when the pattern contains multiple wildcards. For example,
expand("sorted_reads/{sample}.{replicate}.bam", sample=SAMPLES, replicate=[0, 1])
would create the product of all elements of SAMPLES and the list [0, 1], yielding
["sorted_reads/A.0.bam", "sorted_reads/A.1.bam", "sorted_reads/B.0.bam", "sorted_reads/B.1.bam"]
Here, we use only the simple case of expand.
We first let Snakemake know which samples we want to consider.
Remember that Snakemake works top-down, it does not automatically infer this from, e.g., the fastq files in the data folder.
Also remember that Snakefiles are in principle Python code enhanced by some declarative statements to define workflows.
Hence, we can define the list of samples ad-hoc in plain Python at the top of the Snakefile:
SAMPLES = ["A", "B"]
Later, we will learn about more sophisticated ways like config files. Now, we can add the following rule to our Snakefile:
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
With multiple input or output files, it is sometimes handy to refer them separately in the shell command.
This can be done by specifying names for input or output files (here, e.g., fa=...).
The files can then be referred in the shell command via, e.g., {input.fa}.
For long shell commands like this one, it is advisable to split the string over multiple indented lines.
Python will automatically merge it into one.
Further, you will notice that the input or output file lists can contain arbitrary Python statements, as long as it returns a string, or a list of strings.
Here, we invoke our expand function to aggregate over the aligned reads of all samples.
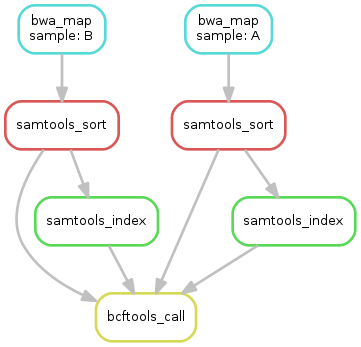
Exercise¶
- obtain the updated DAG of jobs for the target file
calls/all.vcf, it should look like this:
Step 6: Writing a report¶
Although Snakemake workflows are already self-documenting to a certain degree, it is often useful to summarize the obtained results and performed steps in a comprehensive report.
With Snakemake, such reports can be composed easily with the built-in report function.
It is best practice to create reports in a separate rule that takes all desired results as input files and provides a single HTML file as output.
rule report:
input:
"calls/all.vcf"
output:
"report.html"
run:
from snakemake.utils import report
with open(input[0]) as vcf:
n_calls = sum(1 for l in vcf if not l.startswith("#"))
report("""
An example variant calling workflow
===================================
Reads were mapped to the Yeast
reference genome and variants were called jointly with
SAMtools/BCFtools.
This resulted in {n_calls} variants (see Table T1_).
""", output[0], T1=input[0])
First, we notice that this rule does not entail a shell command.
Instead, we use the run directive, which is followed by plain Python code.
Similar to the shell case, we have access to input and output files, which we can handle as plain Python objects (no braces notation here).
We go through the run block line by line.
First, we import the report function from snakemake.utils.
Second, we open the VCF file by accessing it via its index in the input files (i.e. input[0]), and count the number of non-header lines (which is equivalent to the number of variant calls).
Third, we create the report using the report function.
The function takes a string that contains RestructuredText markup.
In addition, we can use the familiar braces notation to access any Python variables (here the samples and n_calls variables we have defined before).
The second argument of the report function is the path were the report will be stored (the function creates a single HTML file).
Then, report expects any number of keyword arguments referring to files that shall be embedded into the report.
Technically, this means that the file will be stored as a Base64 encoded data URI within the HTML file, making reports entirely self-contained.
Importantly, you can refer to the files from within the report via the given keywords followed by an underscore (here T1_).
Hence, reports can be used to semantically connect and explain the obtained results.
When having many result files, it is sometimes handy to define the names already in the list of input files and unpack these into keyword arguments as follows:
report("""...""", output[0], **input)
Further, you can add meta data in the form of any string that will be displayed in the footer of the report, e.g.
report("""...""", output[0], metadata="Author: Johannes Köster (koester@jimmy.harvard.edu)", **input)
Step 7: Adding a target rule¶
So far, we always executed the workflow by specifying a target file at the command line.
Apart from filenames, Snakemake also accepts rule names as targets if the referred rule does not have wildcards.
Hence, it is possible to write target rules collecting particular subsets of the desired results or all results.
Moreover, if no target is given at the command line, Snakemake will define the first rule of the Snakefile as the target.
Hence, it is best practice to have a rule all at the top of the workflow which has all typically desired target files as input files.
Here, this means that we add a rule
rule all:
input:
"report.html"
to the top of our workflow. When executing Snakemake with
$ snakemake -n
the execution plan for creating the file report.html which contains and summarizes all our results will be shown.
Note that, apart from Snakemake considering the first rule of the workflow as default target, the appearance of rules in the Snakefile is arbitrary and does not influence the DAG of jobs.
Exercise¶
- Create the DAG of jobs for the complete workflow.
- Execute the complete workflow and have a look at the resulting
report.htmlin your browser. - Snakemake provides handy flags for forcing re-execution of parts of the workflow. Have a look at the command line help with
snakemake --helpand search for the flag--forcerun. Then, use this flag to re-execute the rulesamtools_sortand see what happens. - With
--reasonit is possible to display the execution reason for each job. Try this flag together with a dry-run and the--forcerunflag to understand the decisions of Snakemake.
Summary¶
In total, the resulting workflow looks like this:
SAMPLES = ["A", "B"]
rule all:
input:
"report.html"
rule bwa_map:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped_reads/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input}"
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
rule report:
input:
"calls/all.vcf"
output:
"report.html"
run:
from snakemake.utils import report
with open(input[0]) as vcf:
n_calls = sum(1 for l in vcf if not l.startswith("#"))
report("""
An example variant calling workflow
===================================
Reads were mapped to the Yeast
reference genome and variants were called jointly with
SAMtools/BCFtools.
This resulted in {n_calls} variants (see Table T1_).
""", output[0], T1=input[0])