Google Life Sciences Tutorial¶
Setup¶
To go through this tutorial, you need the following software installed:
First, you have to install the Miniconda Python3 distribution. See here for installation instructions. Make sure to …
Install the Python 3 version of Miniconda.
Answer yes to the question whether conda shall be put into your PATH.
The default conda solver is a bit slow and sometimes has issues with selecting the latest package releases. Therefore, we recommend to install Mamba as a drop-in replacement via
$ conda install -c conda-forge mamba
Then, you can install Snakemake with
$ mamba create -c conda-forge -c bioconda -n snakemake snakemake
from the Bioconda channel. This will install snakemake into an isolated software environment, that has to be activated with
$ conda activate snakemake
$ snakemake --help
Credentials¶
Using the Google Life Sciences executor with Snakemake requires the environment variable GOOGLE_APPLICATION_CREDENTIALS exported, which should point to the full path of the file on your local machine. To generate this file, you can refer to the page under iam-admin to download your service account key and export it to the environment.
export GOOGLE_APPLICATION_CREDENTIALS="/home/[username]/credentials.json"
The suggested, minimal permissions required for this role include the following:
Compute Storage Admin(Can potentially be restricted further)
Compute Viewer
Service Account User
Cloud Life Sciences Workflows Runner
Service Usage Consumer
Step 1: Upload Your Data¶
We will be obtaining inputs from Google Cloud Storage, as well as saving outputs there. You should first clone the repository with the Snakemake tutorial data:
git clone https://github.com/snakemake/snakemake-lsh-tutorial-data
cd snakemake-lsh-tutorial-data
And then either manually create a bucket and upload data files there, or use the provided script and instructions to do it programatically from the command line. The script generally works like:
python upload_google_storage.py <bucket>/<subpath> <folder>
And you aren’t required to provide a subfolder path if you want to upload to the root of a bucket. As an example, for this tutorial we upload the contents of “data” to the root of the bucket snakemake-testing-data
export GOOGLE_APPLICATION_CREDENTIALS="/path/to/credentials.json"
python upload_google_storage.py snakemake-testing-data data/
If you wanted to upload to a “subfolder” path in a bucket, you would do that as follows:
export GOOGLE_APPLICATION_CREDENTIALS="/path/to/credentials.json"
python upload_google_storage.py snakemake-testing-data/subfolder data/
Your bucket (and the folder prefix) will be referred to as the –default-remote-prefix when you run snakemake. You can visually browse your data in the storage browser <https://console.cloud.google.com/storage/>_.
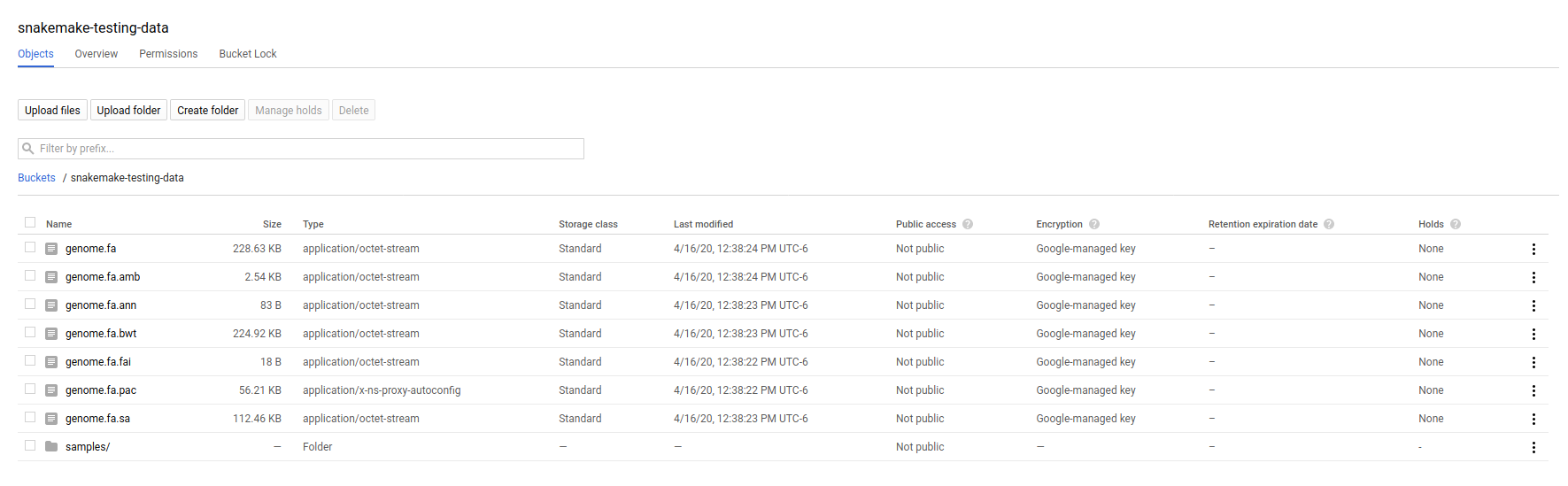
Step 2: Write your Snakefile, Environment File, and Scripts¶
Now that we’ve exported our credentials and have all dependencies installed, let’s get our workflow! This is the exact same workflow from the basic tutorial, so if you need a refresher on the design or basics, please see those pages. You can find the Snakefile, supporting scripts for plotting and environment in the snakemake-lsh-tutorial-data repository.
First, how does a working directory work for this executor? The present working directory, as identified by Snakemake that has the Snakefile, and where a more advanced setup might have a folder of environment specifications (env) a folder of scripts (scripts), and rules (rules), is considered within the context of the build. When the Google Life Sciences executor is used, it generates a build package of all of the files here (within a reasonable size) and uploads those to storage. This package includes the .snakemake folder that would have been generated locally. The build package is then downloaded and extracted by each cloud executor, which is a Google Compute instance.
We next need an environment.yaml file that will define the dependencies that we want installed with conda for our job. If you cloned the “snakemake-lsh-tutorial-data” repository you will already have this, and you are good to go. If not, save this to environment.yaml in your working directory:
channels:
- conda-forge
- bioconda
dependencies:
- python =3.6
- jinja2 =2.10
- networkx =2.1
- matplotlib =2.2.3
- graphviz =2.38.0
- bcftools =1.9
- samtools =1.9
- bwa =0.7.17
- pysam =0.15.0
Notice that we reference this environment.yaml file in the Snakefile below. Importantly, if you were optimizing a pipeline, you would likely have a folder “envs” with more than one environment specification, one for each step. This workflow uses the same environment (with many dependencies) instead of this strategy to minimize the number of files for you.
The Snakefile (also included in the repository) then has the following content. It’s important to note that we have not customized this file from the basic tutorial to hard code any storage. We will be telling snakemake to use the remote bucket as storage instead of the local filesystem.
SAMPLES = ["A", "B"]
rule all:
input:
"plots/quals.svg"
rule bwa_map:
input:
fastq="samples/{sample}.fastq",
idx=multiext("genome.fa", ".amb", ".ann", ".bwt", ".pac", ".sa")
conda:
"environment.yaml"
output:
"mapped_reads/{sample}.bam"
params:
idx=lambda w, input: os.path.splitext(input.idx[0])[0]
shell:
"bwa mem {params.idx} {input.fastq} | samtools view -Sb - > {output}"
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam"
conda:
"environment.yaml"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
conda:
"environment.yaml"
shell:
"samtools index {input}"
rule bcftools_call:
input:
fa="genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
bai=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
conda:
"environment.yaml"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
rule plot_quals:
input:
"calls/all.vcf"
output:
"plots/quals.svg"
conda:
"environment.yaml"
script:
"plot-quals.py"
And make sure you also have the script plot-quals.py in your present working directory for the last step. This script will help us to do the plotting, and is also included in the snakemake-lsh-tutorial-data repository.
import matplotlib
matplotlib.use("Agg")
import matplotlib.pyplot as plt
from pysam import VariantFile
quals = [record.qual for record in VariantFile(snakemake.input[0])]
plt.hist(quals)
plt.savefig(snakemake.output[0])
Step 3: Run Snakemake¶
Now let’s run Snakemake with the Google Life Sciences Executor.
snakemake --google-lifesciences --default-remote-prefix snakemake-testing-data --use-conda --google-lifesciences-region us-west1
The flags above refer to:
–google-lifesciences: to indicate that we want to use the Google Life Sciences API
–default-remote-prefix: refers to the Google Storage bucket. The bucket name is “snakemake-testing-data” and the “subfolder” (or path) (not defined above) would be a subfolder, if needed.
–google-lifesciences-region: the region that you want the instances to deploy to. Your storage bucket should be accessible from here, and your selection can have a small influence on the machine type selected.
Once you submit the job, you’ll immediately see the familiar Snakemake console output, but with additional lines for inspecting google compute instances with gcloud:
Building DAG of jobs...
Unable to retrieve additional files from git. This is not a git repository.
Using shell: /bin/bash
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 all
1 bcftools_call
2 bwa_map
1 plot_quals
2 samtools_index
2 samtools_sort
9
[Thu Apr 16 19:16:24 2020]
rule bwa_map:
input: snakemake-testing-data/genome.fa, snakemake-testing-data/samples/B.fastq
output: snakemake-testing-data/mapped_reads/B.bam
jobid: 8
wildcards: sample=B
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe 13586583122112209762
gcloud beta lifesciences operations list
Take note of those last three lines to describe and list operations - this is how you get complete error and output logs for the run, which we will demonstrate using later.
And you’ll see a block like that for each rule. Here is what the entire workflow looks like after completion:
Building DAG of jobs...
Unable to retrieve additional files from git. This is not a git repository.
Using shell: /bin/bash
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 all
1 bcftools_call
2 bwa_map
1 plot_quals
2 samtools_index
2 samtools_sort
9
[Fri Apr 17 20:27:51 2020]
rule bwa_map:
input: snakemake-testing-data/samples/B.fastq, snakemake-testing-data/genome.fa.amb, snakemake-testing-data/genome.fa.ann, snakemake-testing-data/genome.fa.bwt, snakemake-testing-data/genome.fa.pac, snakemake-testing-data/genome.fa.sa
output: snakemake-testing-data/mapped_reads/B.bam
jobid: 8
wildcards: sample=B
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us-west2/operations/16135317625786219242
gcloud beta lifesciences operations list
[Fri Apr 17 20:31:16 2020]
Finished job 8.
1 of 9 steps (11%) done
[Fri Apr 17 20:31:16 2020]
rule bwa_map:
input: snakemake-testing-data/samples/A.fastq, snakemake-testing-data/genome.fa.amb, snakemake-testing-data/genome.fa.ann, snakemake-testing-data/genome.fa.bwt, snakemake-testing-data/genome.fa.pac, snakemake-testing-data/genome.fa.sa
output: snakemake-testing-data/mapped_reads/A.bam
jobid: 7
wildcards: sample=A
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us-west2/operations/5458247376121133509
gcloud beta lifesciences operations list
[Fri Apr 17 20:34:30 2020]
Finished job 7.
2 of 9 steps (22%) done
[Fri Apr 17 20:34:30 2020]
rule samtools_sort:
input: snakemake-testing-data/mapped_reads/B.bam
output: snakemake-testing-data/sorted_reads/B.bam
jobid: 4
wildcards: sample=B
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us-west2/operations/13750029425473765929
gcloud beta lifesciences operations list
[Fri Apr 17 20:37:34 2020]
Finished job 4.
3 of 9 steps (33%) done
[Fri Apr 17 20:37:35 2020]
rule samtools_sort:
input: snakemake-testing-data/mapped_reads/A.bam
output: snakemake-testing-data/sorted_reads/A.bam
jobid: 3
wildcards: sample=A
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us-west2/operations/15643873965497084056
gcloud beta lifesciences operations list
[Fri Apr 17 20:40:37 2020]
Finished job 3.
4 of 9 steps (44%) done
[Fri Apr 17 20:40:38 2020]
rule samtools_index:
input: snakemake-testing-data/sorted_reads/B.bam
output: snakemake-testing-data/sorted_reads/B.bam.bai
jobid: 6
wildcards: sample=B
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us-west2/operations/6525320566174651173
gcloud beta lifesciences operations list
[Fri Apr 17 20:43:41 2020]
Finished job 6.
5 of 9 steps (56%) done
[Fri Apr 17 20:43:41 2020]
rule samtools_index:
input: snakemake-testing-data/sorted_reads/A.bam
output: snakemake-testing-data/sorted_reads/A.bam.bai
jobid: 5
wildcards: sample=A
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us-west2/operations/9175497885319251567
gcloud beta lifesciences operations list
[Fri Apr 17 20:46:44 2020]
Finished job 5.
6 of 9 steps (67%) done
[Fri Apr 17 20:46:44 2020]
rule bcftools_call:
input: snakemake-testing-data/genome.fa, snakemake-testing-data/sorted_reads/A.bam, snakemake-testing-data/sorted_reads/B.bam, snakemake-testing-data/sorted_reads/A.bam.bai, snakemake-testing-data/sorted_reads/B.bam.bai
output: snakemake-testing-data/calls/all.vcf
jobid: 2
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us-west2/operations/622600526583374352
gcloud beta lifesciences operations list
[Fri Apr 17 20:49:57 2020]
Finished job 2.
7 of 9 steps (78%) done
[Fri Apr 17 20:49:57 2020]
rule plot_quals:
input: snakemake-testing-data/calls/all.vcf
output: snakemake-testing-data/plots/quals.svg
jobid: 1
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us-west2/operations/9350722561866518561
gcloud beta lifesciences operations list
[Fri Apr 17 20:53:10 2020]
Finished job 1.
8 of 9 steps (89%) done
[Fri Apr 17 20:53:10 2020]
localrule all:
input: snakemake-testing-data/plots/quals.svg
jobid: 0
resources: mem_mb=15360, disk_mb=128000
Downloading from remote: snakemake-testing-data/plots/quals.svg
Finished download.
[Fri Apr 17 20:53:10 2020]
Finished job 0.
9 of 9 steps (100%) done
Complete log: /home/vanessa/snakemake-work/tutorial/.snakemake/log/2020-04-17T202749.218820.snakemake.log
We’ve finished the run, great! Let’s inspect our results.
Step 4: View Results¶
The entirety of the log that was printed to the terminal will be available on your local machine where you submit the job in the hidden .snakemake folder under “log” and timestamped accordingly. If you look at the last line in the output above, you’ll see the full path to this file.
You also might notice a line about downloading results:
Downloading from remote: snakemake-testing-data/plots/quals.svg
Since we defined this to be the target of our run
rule all:
input:
"plots/quals.svg"
this fill is downloaded to our host too. Actually, you’ll notice that paths in storage are mirrored on your filesystem (this is what the workers do too):
$ tree snakemake-testing-data/
snakemake-testing-data/
└── plots
└── quals.svg
We can see the result of our run, quals.svg, below:

And if we look at the remote storage, we see that the result file (under plots) and intermediate results (under sorted_reads and calls) are saved there too!
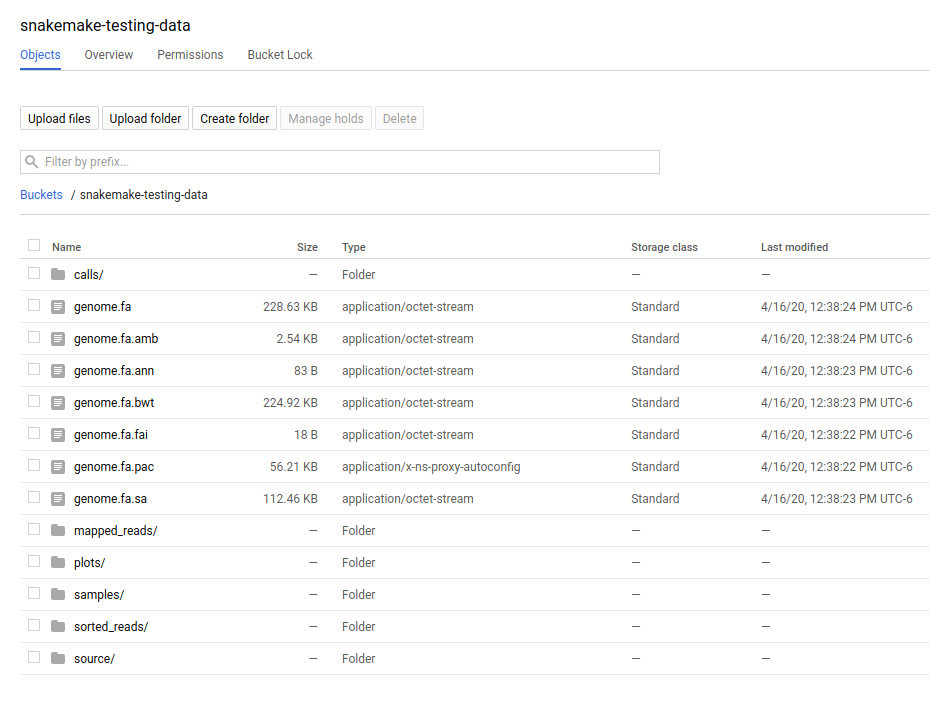
The source folder contains a cache folder with archives that contain your working directories that are extracted on the worker instances. You can safely delete this folder, or keep it if you want to reproduce the run in the future.
Step 5: Debugging¶
Let’s introduce an error (purposefully) into our Snakefile to practice debugging. Let’s remove the conda environment.yaml file for the first rule, so we would expect that Snakemake won’t be able to find the executables for bwa and samtools. In your Snakefile, change this:
rule bwa_map:
input:
fastq="samples/{sample}.fastq",
idx=multiext("genome.fa", ".amb", ".ann", ".bwt", ".pac", ".sa")
conda:
"environment.yaml"
output:
"mapped_reads/{sample}.bam"
params:
idx=lambda w, input: os.path.splitext(input.idx[0])[0]
shell:
"bwa mem {params.idx} {input.fastq} | samtools view -Sb - > {output}"
to this:
rule bwa_map:
input:
fastq="samples/{sample}.fastq",
idx=multiext("genome.fa", ".amb", ".ann", ".bwt", ".pac", ".sa")
output:
"mapped_reads/{sample}.bam"
params:
idx=lambda w, input: os.path.splitext(input.idx[0])[0]
shell:
"bwa mem {params.idx} {input.fastq} | samtools view -Sb - > {output}"
And then for the same command to run everything again, you would need to remove the plots, mapped_reads, and calls folders. Instead, we can make this request more easily by adding the argument –forceall:
snakemake --google-lifesciences --default-remote-prefix snakemake-testing-data --use-conda --google-lifesciences-region us-west1 --forceall
Everything will start out okay as it did before, and it will pause on the first step when it’s deploying the first container image. The last part of the log will look somethig like this:
[Fri Apr 17 22:01:38 2020]
rule bwa_map:
input: snakemake-testing-data/samples/B.fastq, snakemake-testing-data/genome.fa.amb, snakemake-testing-data/genome.fa.ann, snakemake-testing-data/genome.fa.bwt, snakemake-testing-data/genome.fa.pac, snakemake-testing-data/genome.fa.sa
output: snakemake-testing-data/mapped_reads/B.bam
jobid: 8
wildcards: sample=B
resources: mem_mb=15360, disk_mb=128000
Get status with:
gcloud config set project snakemake-testing
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us/operations/11698975339184312706
gcloud beta lifesciences operations list
Since we removed an important dependency to install libraries with conda, we are definitely going to hit an error! That looks like this:
[Fri Apr 17 22:03:08 2020]
Error in rule bwa_map:
jobid: 8
output: snakemake-testing-data/mapped_reads/B.bam
shell:
bwa mem snakemake-testing-data/genome.fa snakemake-testing-data/samples/B.fastq | samtools view -Sb - > snakemake-testing-data/mapped_reads/B.bam
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
jobid: 11698975339184312706
Shutting down, this might take some time.
Oh no! How do we debug it? The error above just indicates that “one of the commands exised with a non-zero exit code,” and that isn’t really enough to know what happened, and how to fix it. Debugging is actually quite simple, we can copy paste the gcloud command to describe our operation into the console. This will spit out an entire structure that shows every step of the rule running, from pulling a container, to downloading the working directory, to running the step.
gcloud beta lifesciences operations describe projects/snakemake-testing/locations/us/operations/11698975339184312706
done: true
error:
code: 9
message: 'Execution failed: generic::failed_precondition: while running "snakejob-bwa_map-8":
unexpected exit status 1 was not ignored'
metadata:
'@type': type.googleapis.com/google.cloud.lifesciences.v2beta.Metadata
createTime: '2020-04-17T22:01:39.642966Z'
endTime: '2020-04-17T22:02:59.149914114Z'
events:
- description: Worker released
timestamp: '2020-04-17T22:02:59.149914114Z'
workerReleased:
instance: google-pipelines-worker-b1cdd36c743c3b477af8114d2511e37e
zone: us-west1-c
- description: 'Execution failed: generic::failed_precondition: while running "snakejob-bwa_map-8":
unexpected exit status 1 was not ignored'
failed:
cause: 'Execution failed: generic::failed_precondition: while running "snakejob-bwa_map-8":
unexpected exit status 1 was not ignored'
code: FAILED_PRECONDITION
timestamp: '2020-04-17T22:02:57.950752682Z'
- description: Unexpected exit status 1 while running "snakejob-bwa_map-8"
timestamp: '2020-04-17T22:02:57.842529458Z'
unexpectedExitStatus:
actionId: 1
exitStatus: 1
- containerStopped:
actionId: 1
exitStatus: 1
stderr: |
me.fa.bwt
Finished download.
/bin/bash: bwa: command not found
/bin/bash: samtools: command not found
[Fri Apr 17 22:02:57 2020]
Error in rule bwa_map:
jobid: 0
output: snakemake-testing-data/mapped_reads/B.bam
shell:
bwa mem snakemake-testing-data/genome.fa snakemake-testing-data/samples/B.fastq | samtools view -Sb - > snakemake-testing-data/mapped_reads/B.bam
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Removing output files of failed job bwa_map since they might be corrupted:
snakemake-testing-data/samples/B.fastq, snakemake-testing-data/genome.fa.amb, snakemake-testing-data/genome.fa.ann, snakemake-testing-data/genome.fa.bwt, snakemake-testing-data/genome.fa.pac, snakemake-testing-data/genome.fa.sa, snakemake-testing-data/mapped_reads/B.bam
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
Complete log: /workdir/.snakemake/log/2020-04-17T220254.129519.snakemake.log
description: |-
Stopped running "snakejob-bwa_map-8": exit status 1: me.fa.bwt
Finished download.
/bin/bash: bwa: command not found
/bin/bash: samtools: command not found
[Fri Apr 17 22:02:57 2020]
Error in rule bwa_map:
jobid: 0
output: snakemake-testing-data/mapped_reads/B.bam
shell:
bwa mem snakemake-testing-data/genome.fa snakemake-testing-data/samples/B.fastq | samtools view -Sb - > snakemake-testing-data/mapped_reads/B.bam
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Removing output files of failed job bwa_map since they might be corrupted:
snakemake-testing-data/samples/B.fastq, snakemake-testing-data/genome.fa.amb, snakemake-testing-data/genome.fa.ann, snakemake-testing-data/genome.fa.bwt, snakemake-testing-data/genome.fa.pac, snakemake-testing-data/genome.fa.sa, snakemake-testing-data/mapped_reads/B.bam
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
Complete log: /workdir/.snakemake/log/2020-04-17T220254.129519.snakemake.log
timestamp: '2020-04-17T22:02:57.842442588Z'
- containerStarted:
actionId: 1
description: Started running "snakejob-bwa_map-8"
timestamp: '2020-04-17T22:02:51.724433437Z'
- description: Stopped pulling "snakemake/snakemake:v5.10.0"
pullStopped:
imageUri: snakemake/snakemake:v5.10.0
timestamp: '2020-04-17T22:02:43.696978950Z'
- description: Started pulling "snakemake/snakemake:v5.10.0"
pullStarted:
imageUri: snakemake/snakemake:v5.10.0
timestamp: '2020-04-17T22:02:10.339950219Z'
- description: Worker "google-pipelines-worker-b1cdd36c743c3b477af8114d2511e37e"
assigned in "us-west1-c"
timestamp: '2020-04-17T22:01:43.232858222Z'
workerAssigned:
instance: google-pipelines-worker-b1cdd36c743c3b477af8114d2511e37e
machineType: n2-highmem-2
zone: us-west1-c
labels:
app: snakemake
name: snakejob-b346c449-9fd6-4f1e-8043-17c300cc9c0d-bwa_map-8
pipeline:
actions:
- commands:
- /bin/bash
- -c
- 'mkdir -p /workdir && cd /workdir && wget -O /download.py https://gist.githubusercontent.com/vsoch/84886ef6469bedeeb9a79a4eb7aec0d1/raw/181499f8f17163dcb2f89822079938cbfbd258cc/download.py
&& chmod +x /download.py && source activate snakemake || true && pip install
crc32c && python /download.py download snakemake-testing-data source/cache/snakeworkdir-5f4f325b9ddb188d5da8bfab49d915f023509c0b1986eb72cb4a2540d7991c12.tar.gz
/tmp/workdir.tar.gz && tar -xzvf /tmp/workdir.tar.gz && snakemake snakemake-testing-data/mapped_reads/B.bam
--snakefile Snakefile --force -j --keep-target-files --keep-remote --latency-wait
0 --attempt 1 --force-use-threads --allowed-rules bwa_map --nocolor --notemp
--no-hooks --nolock --use-conda --default-remote-provider GS --default-remote-prefix
snakemake-testing-data --default-resources "mem_mb=15360" "disk_mb=128000" '
containerName: snakejob-bwa_map-8
imageUri: snakemake/snakemake:v5.10.0
labels:
app: snakemake
name: snakejob-b346c449-9fd6-4f1e-8043-17c300cc9c0d-bwa_map-8
resources:
regions:
- us-west1
virtualMachine:
bootDiskSizeGb: 135
bootImage: projects/cos-cloud/global/images/family/cos-stable
labels:
app: snakemake
goog-pipelines-worker: 'true'
machineType: n2-highmem-2
serviceAccount:
email: default
scopes:
- https://www.googleapis.com/auth/cloud-platform
timeout: 604800s
startTime: '2020-04-17T22:01:43.232858222Z'
name: projects/411393320858/locations/us/operations/11698975339184312706
The log is hefty, so let’s break it into pieces to talk about. Firstly, it’s intended to be read from the bottom up if you want to see things in chronological order. The very bottom line is the unique id of the operation, and this is what you used (with the project identifier string, the number after projects, replaced with your project name) to query for the log. Let’s look at the next section, pipeline. This was the specification built up by Snakemake and sent to the Google Life Sciences API as a request:
pipeline:
actions:
- commands:
- /bin/bash
- -c
- 'mkdir -p /workdir && cd /workdir && wget -O /download.py https://gist.githubusercontent.com/vsoch/84886ef6469bedeeb9a79a4eb7aec0d1/raw/181499f8f17163dcb2f89822079938cbfbd258cc/download.py
&& chmod +x /download.py && source activate snakemake || true && pip install
crc32c && python /download.py download snakemake-testing-data source/cache/snakeworkdir-5f4f325b9ddb188d5da8bfab49d915f023509c0b1986eb72cb4a2540d7991c12.tar.gz
/tmp/workdir.tar.gz && tar -xzvf /tmp/workdir.tar.gz && snakemake snakemake-testing-data/mapped_reads/B.bam
--snakefile Snakefile --force -j --keep-target-files --keep-remote --latency-wait
0 --attempt 1 --force-use-threads --allowed-rules bwa_map --nocolor --notemp
--no-hooks --nolock --use-conda --default-remote-provider GS --default-remote-prefix
snakemake-testing-data --default-resources "mem_mb=15360" "disk_mb=128000" '
containerName: snakejob-bwa_map-8
imageUri: snakemake/snakemake:v5.10.0
labels:
app: snakemake
name: snakejob-b346c449-9fd6-4f1e-8043-17c300cc9c0d-bwa_map-8
resources:
regions:
- us-west1
virtualMachine:
bootDiskSizeGb: 135
bootImage: projects/cos-cloud/global/images/family/cos-stable
labels:
app: snakemake
goog-pipelines-worker: 'true'
machineType: n2-highmem-2
serviceAccount:
email: default
scopes:
- https://www.googleapis.com/auth/cloud-platform
timeout: 604800s
startTime: '2020-04-17T22:01:43.232858222Z'
There is a lot of useful information here. Under resources:
virtualMachine shows the machineType that should correspond to the instance type. You can specify a full name or prefix with –machine-type-prefix or “machine_type” defined under resources for a step. Since we didn’t set any requirements, it chose a reasonable choice for us. This section also shows the size of the boot disk (in GB) and if you added hardware accelerators (GPU) they should show up here too.
regions is the region that the instance was deployed in, which is important to know if you need to specify to run from a particular region. This parameter defalts to regions in the US, and can be modified with the –google-lifesciences-regions parameter.
Under actions you’ll find a few important fields:
imageUri is important to know to see the version of Snakemake (or another container base) that was used. You can customize this with –container-image, and it will default to the latest snakemake.
commands are the commands run to execute the container (also known as the entrypoint). For example, if you wanted to bring up your own instance manually and pull the container defined by imageUri, you could execute the commands to the container (or shell inside and run them interactively) to interactively debug. Notice that the commands ends with a call to snakemake, and shows the arguments that are used. Make sure that this matches your expectation.
The next set of steps pertain to assigning the worker, pulling the container, and starting it. That looks something like this, and it’s fairly straight forward. You can again see that earlier timestamps are on the bottom.
- containerStarted:
actionId: 1
description: Started running "snakejob-bwa_map-8"
timestamp: '2020-04-17T22:02:51.724433437Z'
- description: Stopped pulling "snakemake/snakemake:v5.10.0"
pullStopped:
imageUri: snakemake/snakemake:v5.10.0
timestamp: '2020-04-17T22:02:43.696978950Z'
- description: Started pulling "snakemake/snakemake:v5.10.0"
pullStarted:
imageUri: snakemake/snakemake:v5.10.0
timestamp: '2020-04-17T22:02:10.339950219Z'
- description: Worker "google-pipelines-worker-b1cdd36c743c3b477af8114d2511e37e"
assigned in "us-west1-c"
timestamp: '2020-04-17T22:01:43.232858222Z'
workerAssigned:
instance: google-pipelines-worker-b1cdd36c743c3b477af8114d2511e37e
machineType: n2-highmem-2
zone: us-west1-c
The next section, when the container is stopped, have the meat of the information that we need to debug! This is the step where there was a non-zero exit code.
- containerStopped:
actionId: 1
exitStatus: 1
stderr: |
me.fa.bwt
Finished download.
/bin/bash: bwa: command not found
/bin/bash: samtools: command not found
[Fri Apr 17 22:02:57 2020]
Error in rule bwa_map:
jobid: 0
output: snakemake-testing-data/mapped_reads/B.bam
shell:
bwa mem snakemake-testing-data/genome.fa snakemake-testing-data/samples/B.fastq | samtools view -Sb - > snakemake-testing-data/mapped_reads/B.bam
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Removing output files of failed job bwa_map since they might be corrupted:
snakemake-testing-data/samples/B.fastq, snakemake-testing-data/genome.fa.amb, snakemake-testing-data/genome.fa.ann, snakemake-testing-data/genome.fa.bwt, snakemake-testing-data/genome.fa.pac, snakemake-testing-data/genome.fa.sa, snakemake-testing-data/mapped_reads/B.bam
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
Complete log: /workdir/.snakemake/log/2020-04-17T220254.129519.snakemake.log
description: |-
Stopped running "snakejob-bwa_map-8": exit status 1: me.fa.bwt
Finished download.
/bin/bash: bwa: command not found
/bin/bash: samtools: command not found
[Fri Apr 17 22:02:57 2020]
Error in rule bwa_map:
jobid: 0
output: snakemake-testing-data/mapped_reads/B.bam
shell:
bwa mem snakemake-testing-data/genome.fa snakemake-testing-data/samples/B.fastq | samtools view -Sb - > snakemake-testing-data/mapped_reads/B.bam
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Removing output files of failed job bwa_map since they might be corrupted:
snakemake-testing-data/samples/B.fastq, snakemake-testing-data/genome.fa.amb, snakemake-testing-data/genome.fa.ann, snakemake-testing-data/genome.fa.bwt, snakemake-testing-data/genome.fa.pac, snakemake-testing-data/genome.fa.sa, snakemake-testing-data/mapped_reads/B.bam
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
Complete log: /workdir/.snakemake/log/2020-04-17T220254.129519.snakemake.log
timestamp: '2020-04-17T22:02:57.842442588Z'
Along with seeing the error in stderr, the description key holds the same error. We see what we would have seen if we were running the bwa mem command on our own command line, that the executables weren’t found:
stderr: |
me.fa.bwt
Finished download.
/bin/bash: bwa: command not found
/bin/bash: samtools: command not found
But we shouldn’t be surprised, we on purpose removed the environment file to install them! This is where you would read the error, and respond by updating your Snakefile with a fix.
Step 6: Adding a Log File¶
How might we do better at debugging in the future? The answer is to add a log file for each step, which is where any stderr will be written in the case of failure. For the same step above, we would update the rule to look like this:
rule bwa_map:
input:
fastq="samples/{sample}.fastq",
idx=multiext("genome.fa", ".amb", ".ann", ".bwt", ".pac", ".sa")
output:
"mapped_reads/{sample}.bam"
params:
idx=lambda w, input: os.path.splitext(input.idx[0])[0]
shell:
"bwa mem {params.idx} {input.fastq} | samtools view -Sb - > {output}"
log:
"logs/bwa_map/{sample}.log"
In the above, we would write a log file to storage in a “subfolder” of the snakemake prefix located at “logs/bwa_map.” The log file will be named according to the sample. You could also imagine a flatted structure with a path like logs/bwa_map-{sample}.log. It’s up to you how you want to organize your output. This means that when you see the error appear in your terminal, you can quickly look at this log file instead of resorting to using the gcloud tool. It’s generally good to remember when debugging that:
You should not make assumptions about anything’s existence. Use print statements to verify.
The biggest errors tend to be syntax and/or path errors
If you want to test a different snakemake container, you can use the –container flag.
If the error is especially challenging, set up a small toy example that implements the most basic functionality that you want to achieve.
If you need help, reach out to ask for it! If there is an issue with the Google Life Sciences workflow executor, please open an issue.
It also sometimes helps to take a break from working on something, and coming back with fresh eyes.